tdhf - is a Dirac-Hartree-Fock program developed by Walter R. Johnson
Calculates energies and w.f. of valence electrons. Has Breit + QED corrections
tdhf input
read(5,1000) ident,jmax,jz,nat,nuc,ion,io, in, inf [format(a4,4i8)]
Example Fr 30 87 223 0 0 9 25 1
character*4 $ident = atomic element symbol {e.g., Fr}
$jmax = # of input fields with shell orbital description (see below)
$jz = nuclear charge Z (atomic number for neutrals)
$nat = atomic mass number A ( atomic weight)
$nuc = (unused set to 0)
$ion = (unused set to 0)
$io = Desired relative precision = 10^(-io) - controls convergence criteria
$in = index of the 1-st valence shell in the cards with orbital description (below)
$inf = (0 or 1) 0=closed-shell only
if 1 do a solution for valence electron in the frozen core approximation
Cards with orbital description:
total number $jmax (above)
* ( n(i),kap(i),iof(i),wh(i), i = 1,jmax ) format(3i4,f12.4)
card format : n kappa iof guessed_energy_in_a.u.
* n(i): principal quantun number
* kap(i): angular quantum number kappa
if $guessed_energy >= 0.0 it is calculated internally from the hydrogenic formula
The guessed_energy for valence orbitals better be good, since the program gives some dumb answers if the hydrogenic default is used.
$iof governs some internal step, the mixing weight between previous and next iteration
the first $in cards describe core orbitals the rest $jz -$in are valence orbitals
if $inf was 1 the next input is
$xa xalpha (some mixing parameter)
read(5,*) r0,hh,mm
These are grid parameters
read(5,*) iparm
$iparm = Type of nuclear parameters
if $iparm = 1 rnuc,cnuc,tnuc expected (all in fermis).
if $iparm != 1: cnuc,anuc,b2,b4
For example for Fr the relevant input is
1.00 ! iparm
.00000 6.83430 2.30000 !rnuc,cnuc,tnuc
We mostly use $iparm = 1. Nuclear parameters can be found here (NuclearPropsWRJ.pdf)
In this table tnuc=2.3 fm; rnuc=0.0000 and the relevant parameter is C(fm)
read(5,*) ex
$ex affects amount of exchange in the Hartree model potential for initializing valence+frozen core potential.
Nominal value = 0


 coordinates. As a result of the calculation I would need to store the wave function. If I were to take a very poor grid of 10 points per coordinate, the storage would require $latex 10^{155}$ memory units.
coordinates. As a result of the calculation I would need to store the wave function. If I were to take a very poor grid of 10 points per coordinate, the storage would require $latex 10^{155}$ memory units.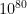 . So even if we were able to compute the Cs wave function, there is no plausible way to store it in usable form.
. So even if we were able to compute the Cs wave function, there is no plausible way to store it in usable form.